Radius of gyration change $\Delta R_g$ due to the ion cloud of a charged protein
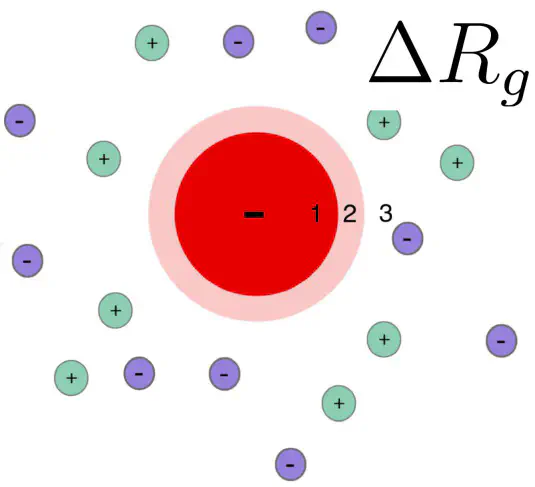
Introduction: influence of the ion cloud of proteins on a SAXS curve
SAXS curves report on the overall electron density contrast of a biomolecule with respect to the bulk solvent. The (counter) ion cloud of charged proteins contributes to the electron density contrast, leading to a modified radius of gyration.
Here, we provide a Python script that estimates the influence of the ion cloud of a protein on the radius of gyration, as derived by a Guinier analysis of SAXS data. The model describes the protein as a uniform sphere, such that the volume, electron density, and the forward scattering intensity of the sphere matches with the protein. Distribution of the ions is calculated with linearize Poisson-Boltzmann theory. For more details, please see our paper. An implementation of the spherical model in Python is provided here: delta-Rg-ioncloud.py
Note: The water density in the hydration layer of biomolecules typically differs from the density of the bulk water, thereby also leading to a modified (often increased) radius of gyration. This phenomenon is not captured by this script. Instead, this script only computes the additional effect of the ion cloud on Rg. If you are interested in the hydration layer effect on Rg, please note our work on explicit-solvent SAXS calculations based on MD simulations, or visit our web server WAXSiS at http://waxsis.uni-saarland.de
If you use this tool for a publication, please cite:
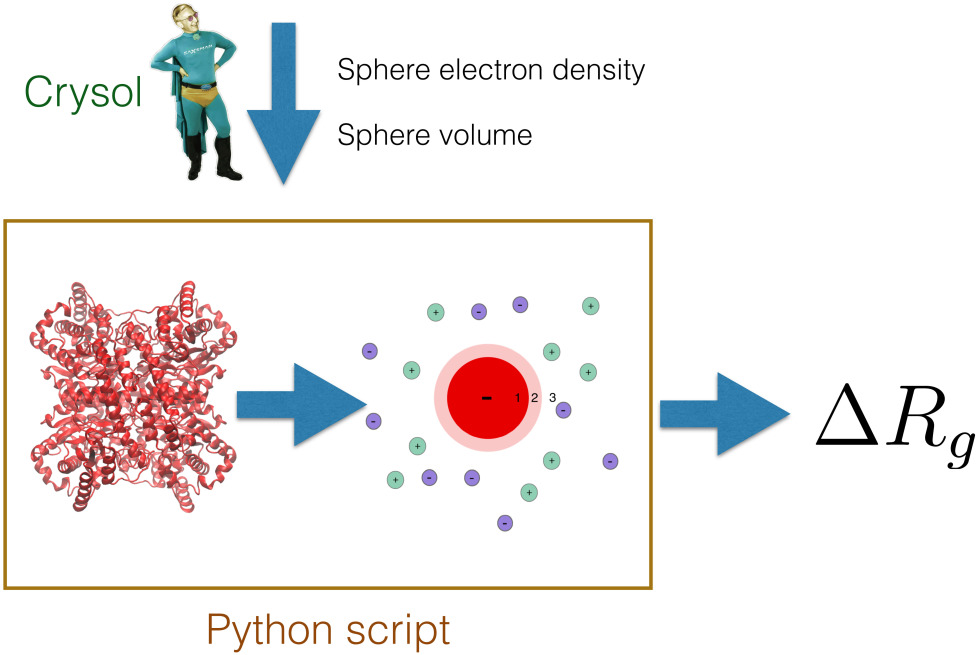
What you need
Two mandatory inputs for the Python scripts are electron density and volume of a biomolecule. These values can be given in a command line, and no additional software is necessary. However, to obtain best estimates, you should provide a PDB file of the biomolecule. Then, the script will use the PDB and the CRYSOL software (part of ATSAS) to estimate the electron density and the volume of the sphere, such that the forward scattering I(q)=0 (or, equivalently, the total contrast) matches with the CRYSOL estimate. For this, you should have Crysol installed on your system.
How to run a script examples
There are two ways to calculate ΔRg using this tool. We demonstrate them for glucose isomerase in a 100mM CsCl solution:
Option 1 (recommended)
Sphere density taken as a density of a biomolecule calculated using Crysol. Volume of the biomolecule calculated from the zero angle scattering (aso taken from Crysol ) and sphere density.
For this you need a protein pdb file. Visit the Protein Data Bank web site and search for 1MNZ.
Download the biological assembly.
Remove organic molecules from the crystallization buffer: egrep -v 'TRS|MRD' 1mnz.pdb > gi.pdb.
Then, run the Python script:
python ./delta-Rg-ioncloud.py --positive_ion_name Cs --negative_ion_name Cl --conc 100 --charge -60 --pdb gi.pdb
You should get ΔRg ~ 0.20 nm.
Option 2
Both the sphere density and the sphere volume taken from a command line. (PDB file and Crysol installation are not required):
python ./delta-Rg-ioncloud.py --positive_ion_name Cs --negative_ion_name Cl --conc 100 --charge -60 --sphere_volume 232.6 --sphere_dens 431.6
You should get ΔRg ~ 0.20 nm.
Every successful run of a script will write 3 files: (i) log file that contains ΔRg estimate, as well as the modelled parameters of the sphere, (ii) file that contains the electron density contrast of the system plotted as a function of distance from the center of the sphere, and (iii) the number densities of anion and cation plotted as a function of distance from the surface of the sphere.
To see more options, please type:
python ./delta-Rg-ioncloud.py --help