Updates
Software

BindFlow
BindFlow is a snakemake-based workflow for FEP and MM(PB/GB)SA calculations with GROMACS.
GROMACS Chain Coordinate
A modification of GROMACS that implements the chain coordinate, a reaction coordinate for pore formation in membranes and stalk formation between membranes.
GROMACS-SWAXS
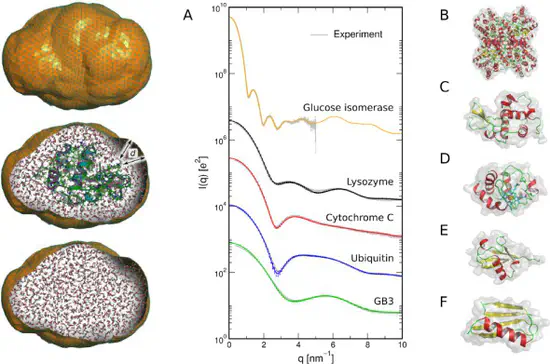
Our modification of GROMACS for computing small- and wide-angle X-ray or neutron scattering curves (SAXS/SANS), small-angle neutron scattering curves (SANS), and for doing SAXS/SANS-driven molecular dynamics simulations.

moldrug
moldrug is a Python package for drug-oriented optimization in the chemical space. It uses a Genetic Algorithm (GA) as a search engine in the chemical space and CReM library as a chemical structure generator.
WAXSiS
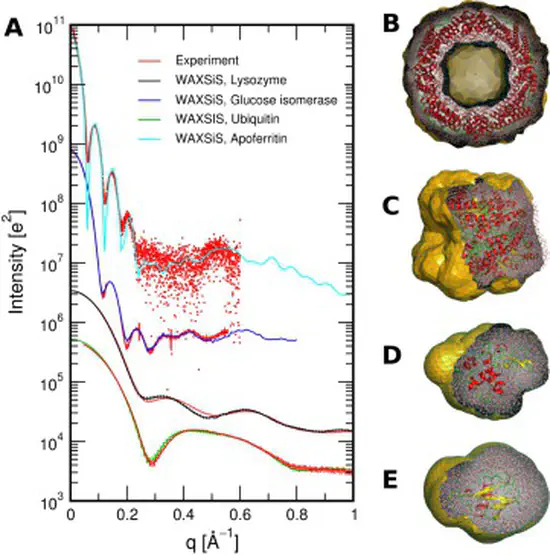
We have developed a web sever called WAXSiS, that computes small- and wide-angle X-ray scattering (SAXS/WAXS) patterns based on explicit-solvent molecular dynamics (MD) simulations. In contrast to previously available methods, WAXSiS provides a highly accurate model for the hydration layer and excluded solvent, and it accounts for thermal fluctuations.
MemGen
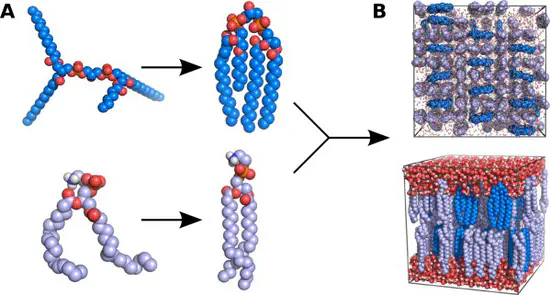
We have developed a new web sever called MemGen, that sets up simulation systems of lipid membranes. MemGen is not restricted to specific force fields, lipid types, or MD simulation software.
Auxiliary
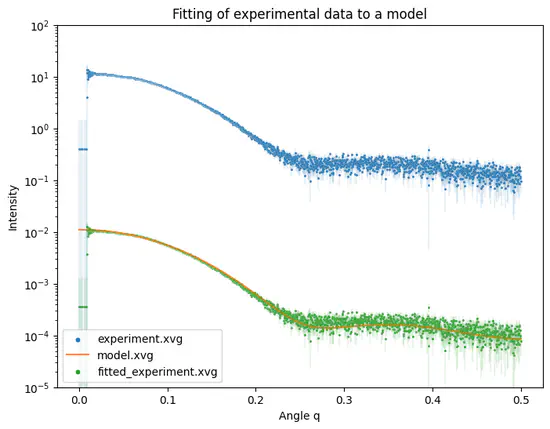
Fit SAS experiment
Python script fit-experiment developed for WAXSiS allows fitting of experimental SAS intensity curves to modelled (calculated/simulated) intensity curves.
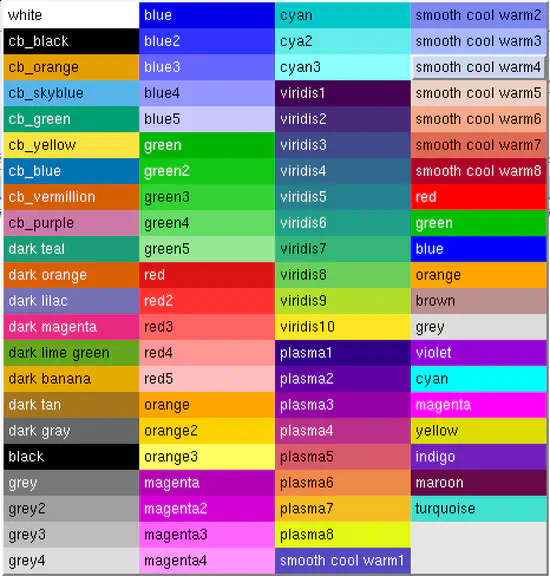
xmgrace colors
xmgrace is versatile 2D plotting tool. Just the default colors are quite ugly. Here, you find a xmgrace default file with many more defined colors. The colors starting with cb_ are recommended to allow color blind people to understand your scientific plots.
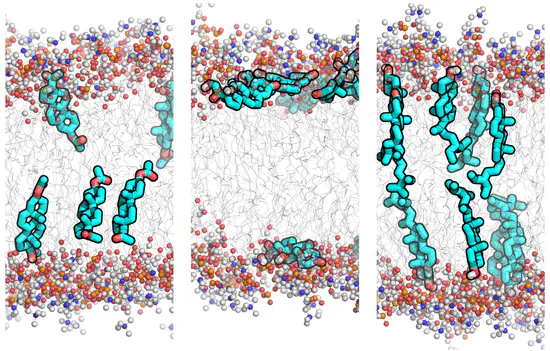
GROMACS GAFF topologies for 26 steroid compounds
Simulation parameters (topologies) for the General Amber Force Field (GAFF) of 26 typical steroid compounds were derived and refined against experimental membrane/water partition coefficients. On this site, we provide force field topology files in GROMACS format for 26 steroid compounds.
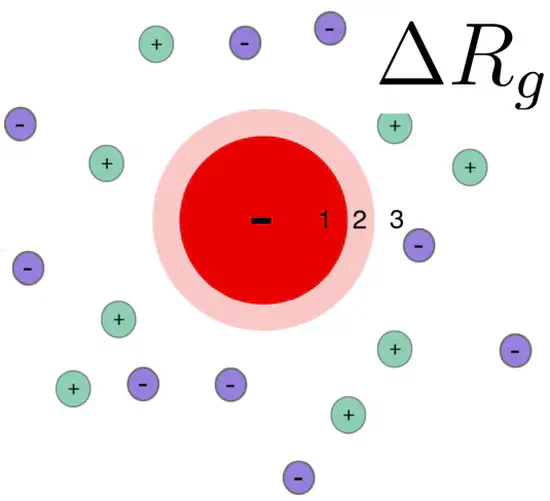
Radius of gyration change $\Delta R_g$ due to the ion cloud of a charged protein
Introduction: influence of the ion cloud of proteins on a SAXS curve SAXS curves report on the overall electron density contrast of a biomolecule with respect to the bulk solvent. The (counter) ion cloud of charged proteins contributes to the electron density contrast, leading to a modified radius of gyration.
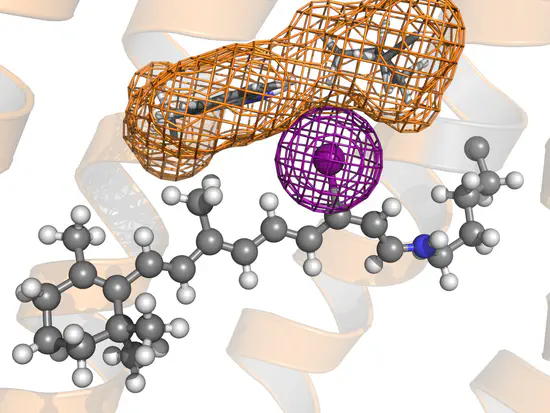
GROMACS topology for retinal
Gromacs topology of a retinal bound to lysine via protonated Schiff base Here you find the files required to use gmx pdb2gmx to generate a GROMACS topology of retinal bound to lysine via a protonated Schiff base.
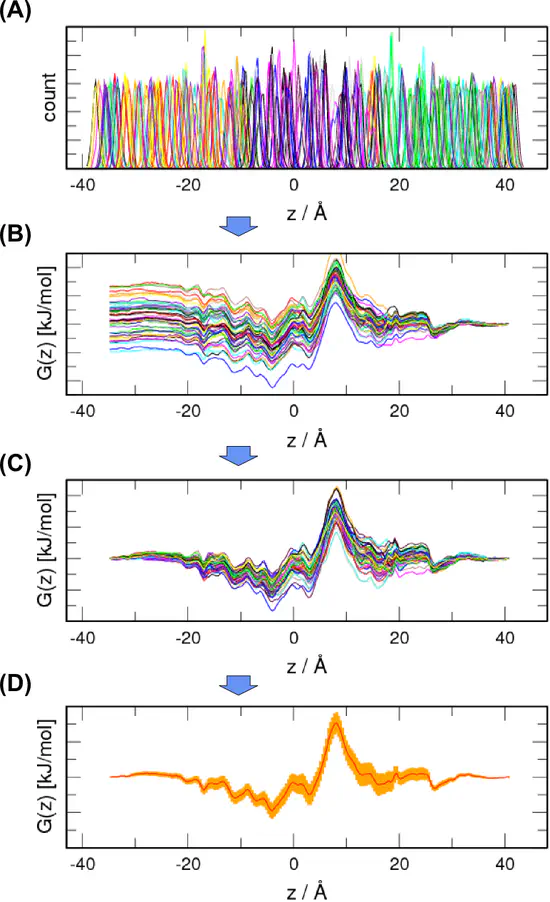
WHAM
The Weighted Histogram Analysis Method (WHAM) is a standard technique used to compute potentials of mean force (PMFs) from a set of umbrella sampling simulations.
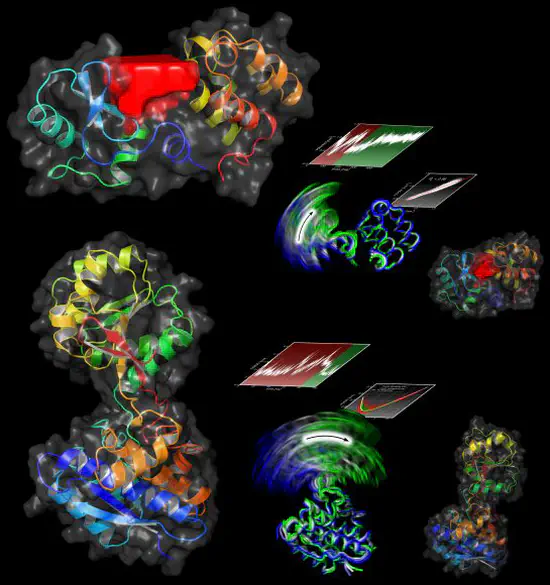
Functional Mode Analysis
We have developed a new technique called Functional Mode Analysis (FMA) that detects collective motions in biomolecules related to a specific function of the biomolecule. Given a large set to structures of one protein, for example from a molecular dynamics trajectory, the method detects a collective motion (or collective mode), that is maximally correlated to an arbitrary quantity of interest.
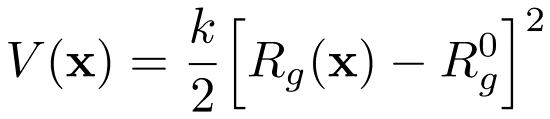
Pulling the Radius of Gyration with GROMACS
We have written a GROMACS extension that allows you to apply a harmonic (or umbrella-like) restraint along the radius of gyration $R_g$ of a group of atoms (typically molecule).