Hydration shells of globular and intrinsically disordered proteins: effects of amino acid composition, peptide conformation, and force fields
Abstract
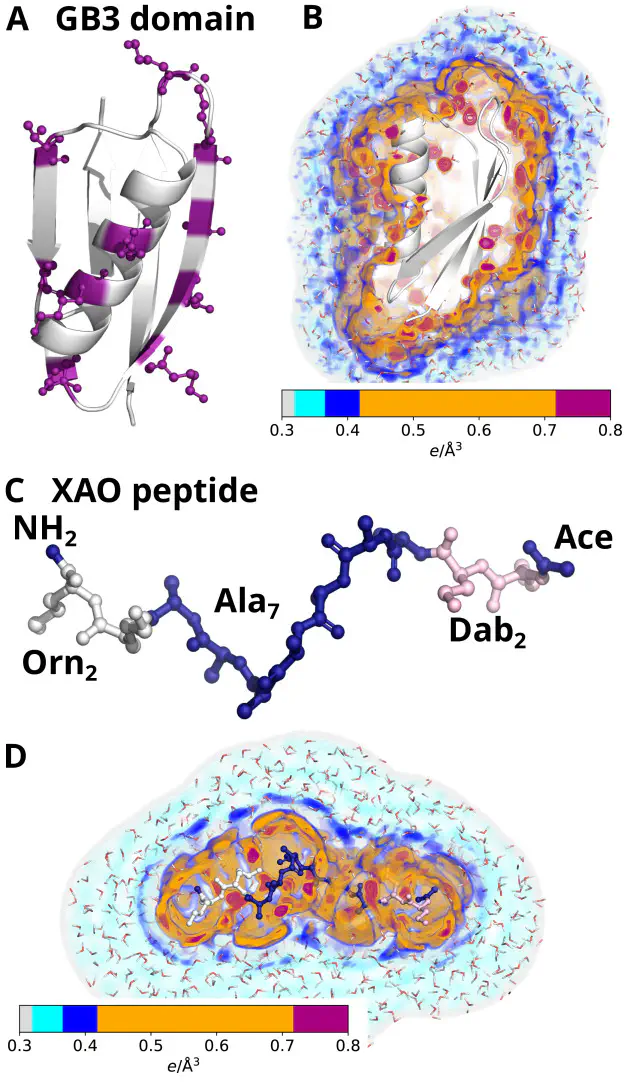
The protein hydration shell is a key mediator of processes such as molecular recognition, protein folding, and proton transfer. How surface-exposed amino acids shape the hydration shell structure is not well understood. We combine molecular dynamics simulations with explicit-solvent predictions of small-angle X-ray scattering (SAXS) curves to quantify the contributions of all 20 proteinogenic amino acids to the hydration shell of the globular GB3 domain and the intrinsically disordered protein (IDP) XAO. We focus on two quantities encoded by SAXS curves: the hydration shell effect on the radius of gyration and the electron density contrast between protein and solvent. We derive an amino-acid-specific contrast score, revealing that acidic residues generate the strongest contrast with 1 to 1.5 excess water molecules relative to alanine, followed by cationic and polar residues. In contrast, apolar residues generate a water depletion layer. These trends are consistent across simulations with different water models. Around the XAO peptide, the hydration shell is generally far weaker compared to the globular GB3 domain, indicating unfavorable water–peptide packing at the IDP surface. The hydration shell effect on the radius of gyration of the IDP is strongly conformation-dependent. Together, the calculations show that the composition and spatial arrangement of surface-exposed amino acids govern the hydration shell structure, with implications for a wide range of biological functions and for hydration-sensitive experimental techniques such as solution scattering.