Molecular simulations reveal the free energy landscape and transition state of membrane electroporation
Abstract
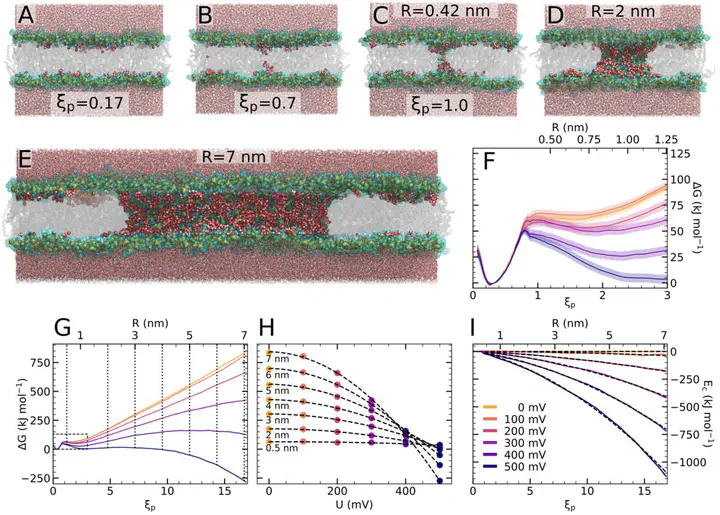
The formation of pores over lipid membranes by the application of electric fields, termed membrane electroporation, is widely used in biotechnology and medicine to deliver drugs, vaccines, or genes into living cells. Continuum models for describing the free energy landscape of membrane electroporation have been proposed decades ago, but they have never been tested against spatially detailed atomistic models. Using molecular dynamics (MD) simulations with a recently proposed reaction coordinate, we computed potentials of mean force of pore nucleation and pore expansion in lipid membranes at various transmembrane potentials. Whereas the free energies of pore expansion are compatible with previous continuum models, the experimentally important free energy barrier of pore nucleation is at variance with established models. We trace the discrepancy to previously incorrect assumptions on the geometry of the transition state; previous continuum models assumed the presence of a membrane-spanning defect throughout the process whereas, according to the MD simulations, the transition state of pore nucleation is typically passed before a transmembrane defect has formed. A modified continuum model is presented that qualitatively agrees with the MD simulations. Using kinetics of pore opening together with transition state theory, our free energies of pore nucleation are in excellent agreement with previous experimental data.