Structure and ensemble refinement against SAXS data: Combining MD simulations with Bayesian inference or with the maximum entropy principle
Abstract
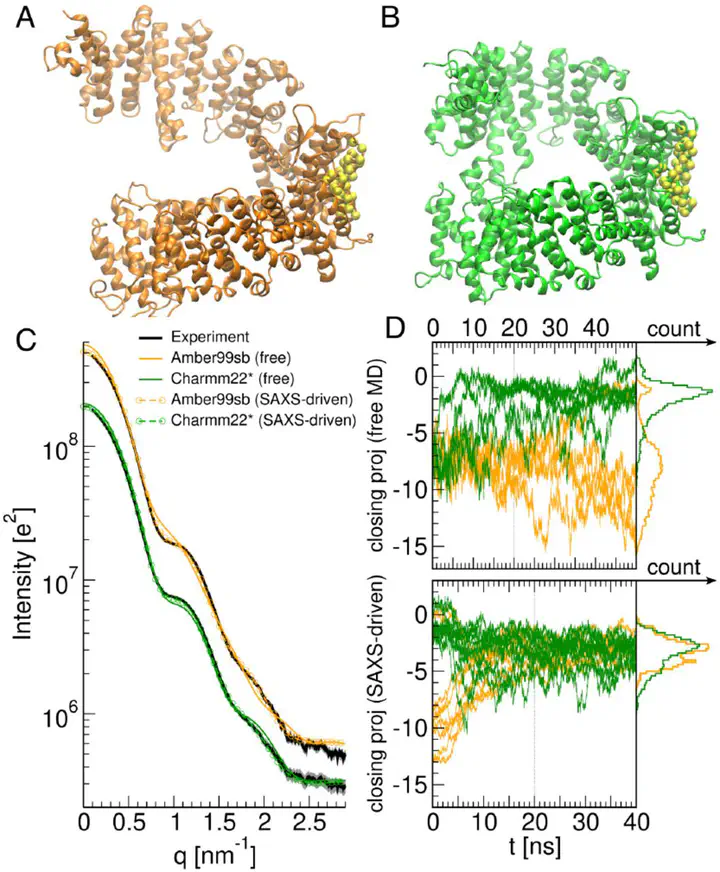
Small-angle X-ray scattering (SAXS) is a powerful method for tracking conformational transitions of proteins or soft-matter complexes in solution. However, the interpretation of the experimental data is challenged by the low spatial resolution and the low information content of the data, which lead to a high risk of overinterpreting the data. Here, we illustrate how SAXS data can be integrated into all-atom molecular dynamics (MD) simulation to derive atomic structures or heterogeneous ensembles that are compatible with the data. Besides providing atomistic insight, the MD simulation adds physicochemical information, as encoded in the MD force fields, which greatly reduces the risk of overinterpretation. We present an introduction into the theory of SAXS-driven MD simulations as implemented in GROMACS-SWAXS, a modified version of the GROMACS simulation software. We discuss SAXS-driven parallel-replica ensemble refinement with commitment to the maximum entropy principle as well as a Bayesian formulation of SAXS-driven structure refinement. Practical considerations for running and interpreting the simulations are presented. The methods are freely available via GitLab at https://gitlab.com/cbjh/gromacs-swaxs.