A round-robin approach provides a detailed assessment of biomolecular small-angle scattering data reproducibility and yields consensus curves for benchmarking
Abstract
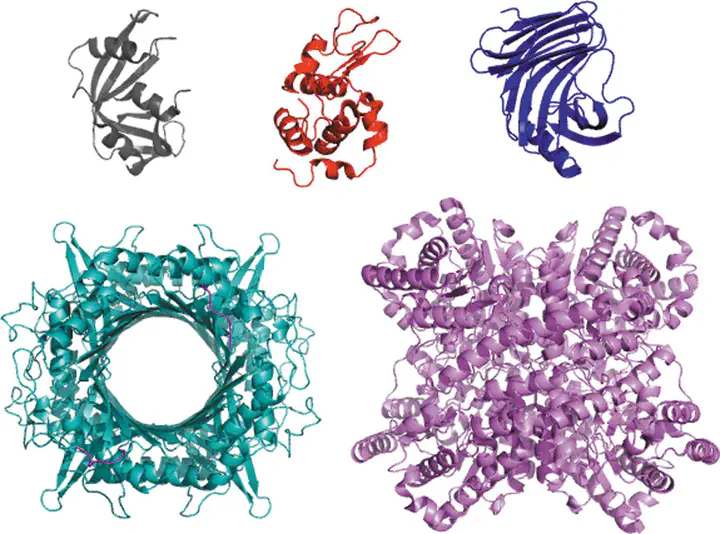
Through an expansive international effort that involved data collection on 12 small-angle X-ray scattering (SAXS) and four small-angle neutron scattering (SANS) instruments, 171 SAXS and 76 SANS measurements for five proteins (ribonuclease A, lysozyme, xylanase, urate oxidase and xylose isomerase) were acquired. From these data, the solvent-subtracted protein scattering profiles were shown to be reproducible, with the caveat that an additive constant adjustment was required to account for small errors in solvent subtraction. Further, the major features of the obtained consensus SAXS data over the q measurement range 0–1 Å−1 are consistent with theoretical prediction. The inherently lower statistical precision for SANS limited the reliably measured q-range to <0.5 Å−1, but within the limits of experimental uncertainties the major features of the consensus SANS data were also consistent with prediction for all five proteins measured in H2O and in D2O. Thus, a foundation set of consensus SAS profiles has been obtained for benchmarking scattering-profile prediction from atomic coordinates. Additionally, two sets of SAXS data measured at different facilities to q > 2.2 Å−1 showed good mutual agreement, affirming that this region has interpretable features for structural modelling. SAS measurements with inline size-exclusion chromatography (SEC) proved to be generally superior for eliminating sample heterogeneity, but with unavoidable sample dilution during column elution, while batch SAS data collected at higher concentrations and for longer times provided superior statistical precision. Careful merging of data measured using inline SEC and batch modes, or low- and high-concentration data from batch measurements, was successful in eliminating small amounts of aggregate or interparticle interference from the scattering while providing improved statistical precision overall for the benchmarking data set.