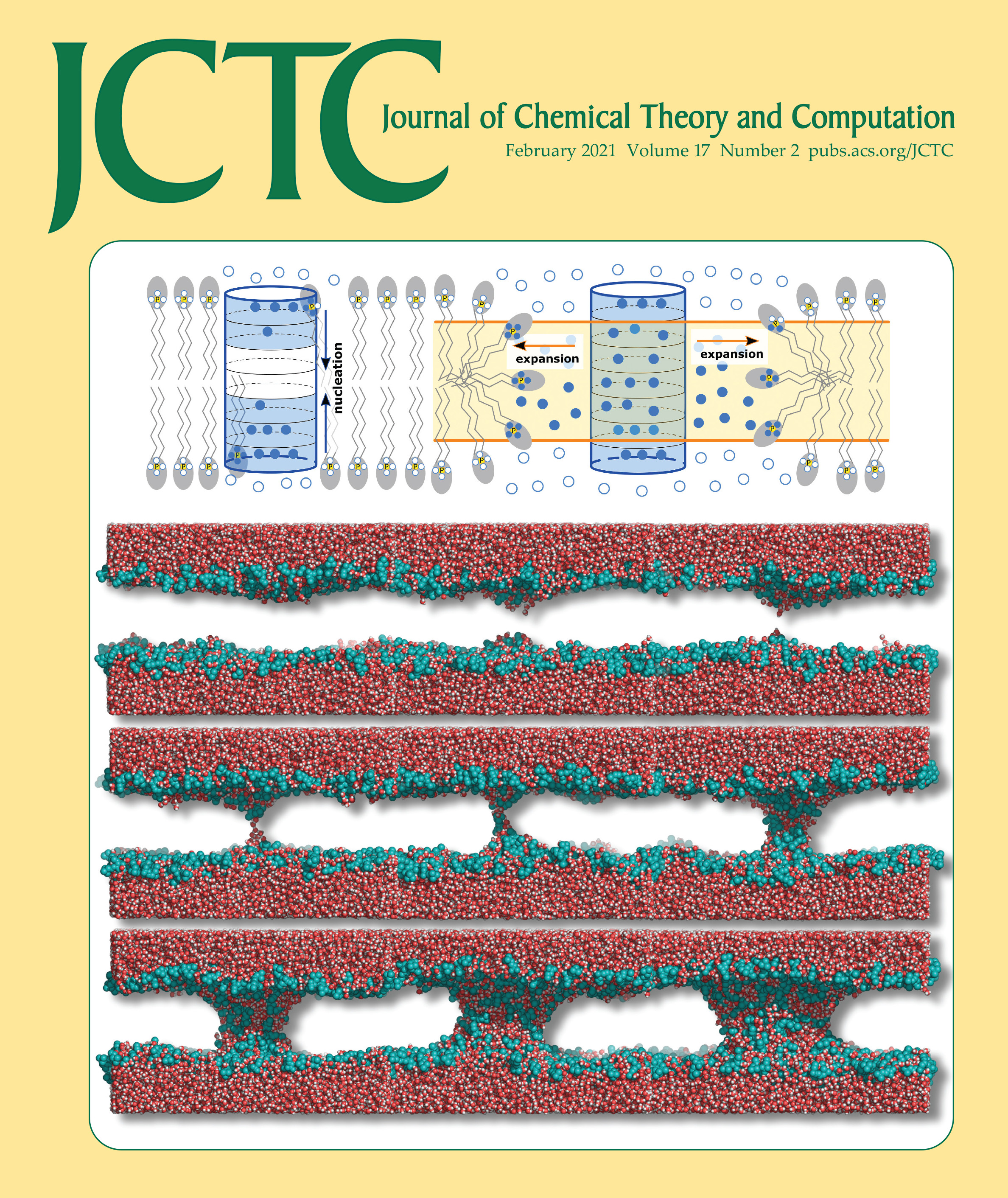
Joint Reaction Coordinate for Computing the Free-Energy Landscape of Pore Nucleation and Pore Expansion in Lipid Membranes
Abstract
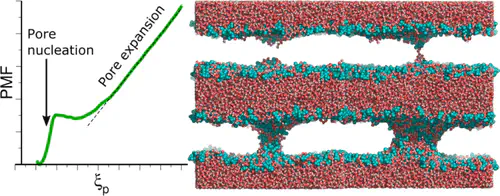
Topological transitions of membranes, such as pore formation or membrane fusion, play key roles in biology, biotechnology, and in medical applications. Calculating the related free-energy landscapes has been complicated by the fact that such processes involve a sequence of transitions along highly distinct directions in conformational space, making it difficult to define good reaction coordinates (RCs) for the overall process. In this study, a new RC capable of driving both pore nucleation and pore expansion in lipid membranes is presented. The potential of mean force (PMF) along the RC computed with molecular dynamics simulations provides a comprehensive view on the free-energy landscape of pore formation, including a barrier for pore nucleation; the size, free energy, and metastability of the open pore; and the energetic cost for further pore expansion against the line tension of the pore rim. The RC is illustrated by quantifying the effects of (i) simulation system size and (ii) the addition of dimethyl sulfoxide on the free-energy landscape of pore formation. PMF calculations along the RC provide mechanistic and energetic understanding of pore formation, hence they will be useful to rationalize the effects of membrane-active peptides, electric fields, and membrane composition on transmembrane pores.