Abstract
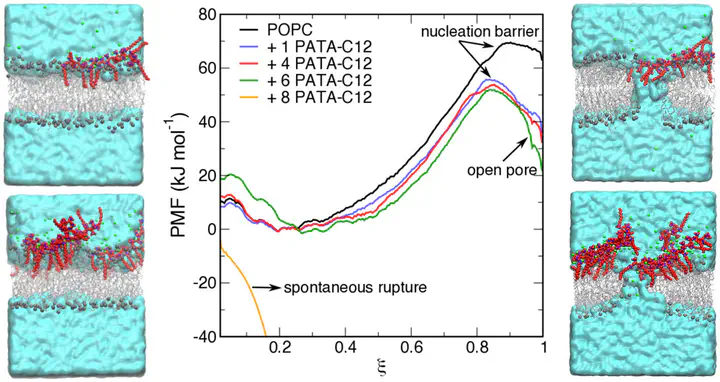
Polycations are an attractive class of macromolecules with promising applications as drug/gene carriers and biocides. The chemical structure and concentration of a polycation determine its interaction with cellular membranes and, hence, are crucial parameters for designing efficient nontoxic polycations. However, the interaction of polycations with biomembranes at the molecular level and the corresponding free-energy landscape is not well understood. In this work, we investigate the molecular mechanism of interaction between a strong polycation substituted with alkyl moieties and zwitterionic membranes via long-time-scale all-atom molecular dynamics simulations and free-energy calculations combined with Langmuir monolayer, atomic force microscopy, and calcein-release experimental measurements. We found that the membrane activity of the polycation and its ability to induce pores in the membranes can be attributed to the polycation-induced changes in the bilayer organization, such as reduced membrane thickness, increased disorder of the acyl chains, reduced packing, and electrostatic field gradients between membrane leaflets. These changes facilitate the penetration of water into the membrane and the formation of aqueous defects/pores. The calculated free-energy profiles indicate that the polycation lowers the nucleation barrier for pore opening and the free energy for pore formation in a concentration-dependent manner. Above the critical coverage of the membrane, the polycation nucleates spontaneous pores in zwitterionic membranes. Our work demonstrates the potential of combining enhanced sampling methods in MD simulations with experiments for a quantitative description of various events in the polycation-membrane interaction cycle, such as strong adsorption on the membrane due to hydrophobic and electrostatic interactions, and pore formation.