Assigning crystallographic electron densities with free energy calculations—The case of the fluoride channel Fluc
Abstract
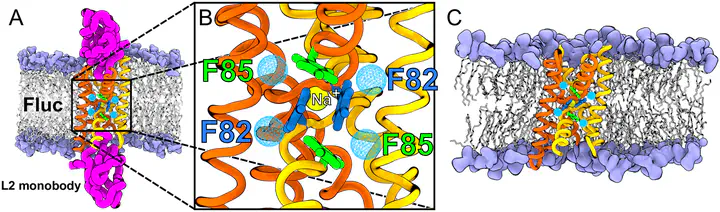
Approximately 90% of the structures in the Protein Data Bank (PDB) were obtained by X-ray crystallography or electron microscopy. Whereas the overall quality of structure is considered high, thanks to a wide range of tools for structure validation, uncertainties may arise from density maps of small molecules, such as organic ligands, ions or water, which are non-covalently bound to the biomolecules. Even with some experience and chemical intuition, the assignment of such disconnected electron densities is often far from obvious. In this study, we suggest the use of molecular dynamics (MD) simulations and free energy calculations, which are well-established computational methods, to aid in the assignment of ambiguous disconnected electron densities. Specifically, estimates of (i) relative binding affinities, for instance between an ion and water, (ii) absolute binding free energies, i.e., free energies for transferring a solute from bulk solvent to a binding site, and (iii) stability assessments during equilibrium simulations may reveal the most plausible assignments. We illustrate this strategy using the crystal structure of the fluoride specific channel (Fluc), which contains five disconnected electron densities previously interpreted as four fluoride and one sodium ion. The simulations support the assignment of the sodium ion. In contrast, calculations of relative and absolute binding free energies as well as stability assessments during free MD simulations suggest that four of the densities represent water molecules instead of fluoride. The assignment of water is compatible with the loss of these densities in the non-conductive F82I/F85I mutant of Fluc. We critically discuss the role of the ion force fields for the calculations presented here. Overall, these findings indicate that MD simulations and free energy calculations are helpful tools for modeling water and ions into crystallographic density maps.