Probing a Continuous Polar Defect: A Reaction Coordinate for Pore Formation in Lipid Membranes
Abstract
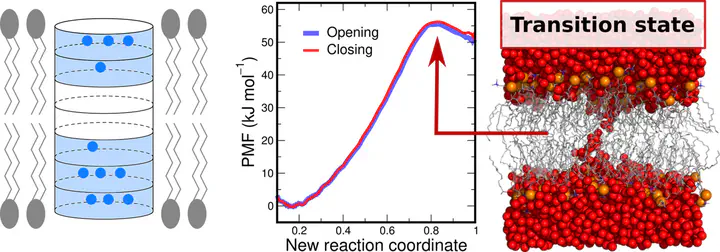
Various biophysical processes involve the formation of aqueous pores over lipid membranes, including processes of membrane fusion, antimicrobial peptide activity, lipid flip-flop, and membrane permeation. Reliable and efficient free-energy calculations of pore formation using molecular dynamics simulations remained challenging due to the lack of good reaction coordinates (RCs) for pore formation. We present a new RC for pore formation that probes the formation and rupture of a continuous polar defect over the membrane. Potential of mean force (PMF) calculations along the new RC rapidly converge and exhibit no hysteresis between pore-opening and pore-closing pathways, in contrast to calculations based on previous RCs. We show that restraints along the new RC may restrain the system tightly to the transition state of pore formation, rationalizing the absence of hysteresis. We observe that the PMF of pore formation in a tension-free membrane of dimyristoylphosphatidylcholine (DMPC) reveals a free-energy barrier for pore nucleation, confirming a long-hypothesized metastable prepore state. We test the influence of the lipid force field, the cutoff distance used for Lennard-Jones interactions, and the lateral membrane size on the free energies of pore formation. In contrast to PMF calculations based on previous RCs, we find that such parameters have a relatively small influence on the free energies of pore nucleation. However, the metastability of the open pore in DMPC may depend on such parameters. The RC has been implemented into an extension of the GROMACS simulation software. The new RC allows for reliable and computationally efficient free-energy calculations of pore formation in lipid membranes.