Interpretation of Solution X-Ray Scattering by Explicit-Solvent Molecular Dynamics
Abstract
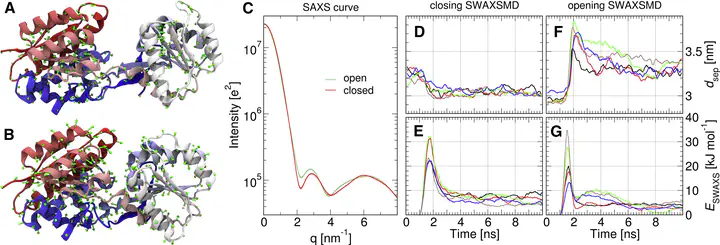
Small- and wide-angle x-ray scattering (SWAXS) and molecular dynamics (MD) simulations are complementary approaches that probe conformational transitions of biomolecules in solution, even in a time-resolved manner. However, the structural interpretation of the scattering signals is challenging, while MD simulations frequently suffer from incomplete sampling or from a force-field bias. To combine the advantages of both techniques, we present a method that incorporates solution scattering data as a differentiable energetic restraint into explicit-solvent MD simulations, termed SWAXS-driven MD, with the aim to direct the simulation into conformations satisfying the experimental data. Because the calculations fully rely on explicit solvent, no fitting parameters associated with the solvation layer or excluded solvent are required, and the calculations remain valid at wide angles. The complementarity of SWAXS and MD is illustrated using three biological examples, namely a periplasmic binding protein, aspartate carbamoyltransferase, and a nuclear exportin. The examples suggest that SWAXS-driven MD is capable of refining structures against SWAXS data without foreknowledge of possible reaction paths. In turn, the SWAXS data accelerates conformational transitions in MD simulations and reduces the force-field bias.
Biophysical Journal Paper of the Year Award
Related New and Notable comment by Robert Rambo and John Tainer: Biophys. J. 108:2421-2423 (2015).