Force Field Benchmark of Organic Liquids: Density, Enthalpy of Vaporization, Heat Capacities, Surface Tension, Isothermal Compressibility, Volumetric Expansion Coefficient, and Dielectric Constant
Abstract
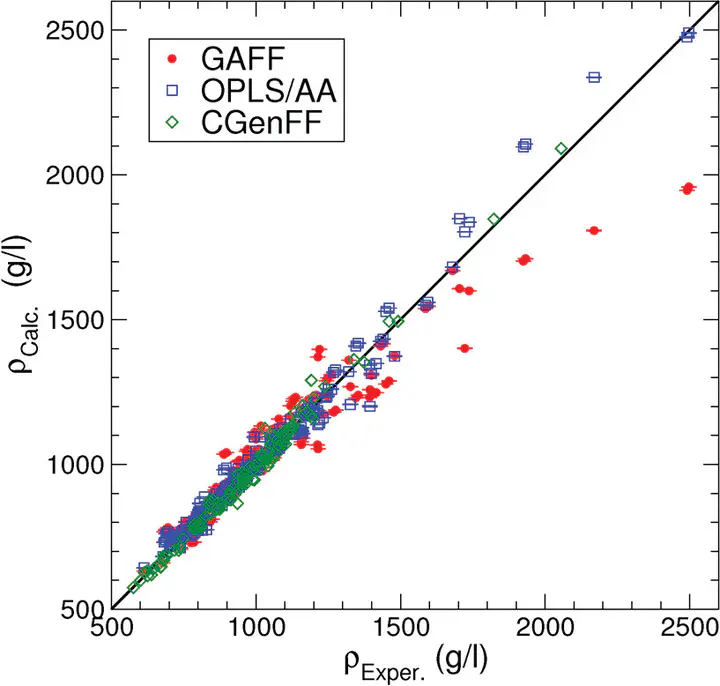
The chemical composition of small organic molecules is often very similar to amino acid side chains or the bases in nucleic acids, and hence there is no a priori reason why a molecular mechanics force field could not describe both organic liquids and biomolecules with a single parameter set. Here, we devise a benchmark for force fields in order to test the ability of existing force fields to reproduce some key properties of organic liquids, namely, the density, enthalpy of vaporization, the surface tension, the heat capacity at constant volume and pressure, the isothermal compressibility, the volumetric expansion coefficient, and the static dielectric constant. Well over 1200 experimental measurements were used for comparison to the simulations of 146 organic liquids. Novel polynomial interpolations of the dielectric constant (32 molecules), heat capacity at constant pressure (three molecules), and the isothermal compressibility (53 molecules) as a function of the temperature have been made, based on experimental data, in order to be able to compare simulation results to them. To compute the heat capacities, we applied the two phase thermodynamics method (Lin et al. J. Chem. Phys.2003, 119, 11792), which allows one to compute thermodynamic properties on the basis of the density of states as derived from the velocity autocorrelation function. The method is implemented in a new utility within the GROMACS molecular simulation package, named g_dos, and a detailed exposé of the underlying equations is presented. The purpose of this work is to establish the state of the art of two popular force fields, OPLS/AA (all-atom optimized potential for liquid simulation) and GAFF (generalized Amber force field), to find common bottlenecks, i.e., particularly difficult molecules, and to serve as a reference point for future force field development. To make for a fair playing field, all molecules were evaluated with the same parameter settings, such as thermostats and barostats, treatment of electrostatic interactions, and system size (1000 molecules). The densities and enthalpy of vaporization from an independent data set based on simulations using the CHARMM General Force Field (CGenFF) presented by Vanommeslaeghe et al. (J. Comput. Chem.2010, 31, 671) are included for comparison. We find that, overall, the OPLS/AA force field performs somewhat better than GAFF, but there are significant issues with reproduction of the surface tension and dielectric constants for both force fields.