Accelerating ligand discovery by combining Bayesian optimization with MMGBSA-based binding affinity calculations
Abstract
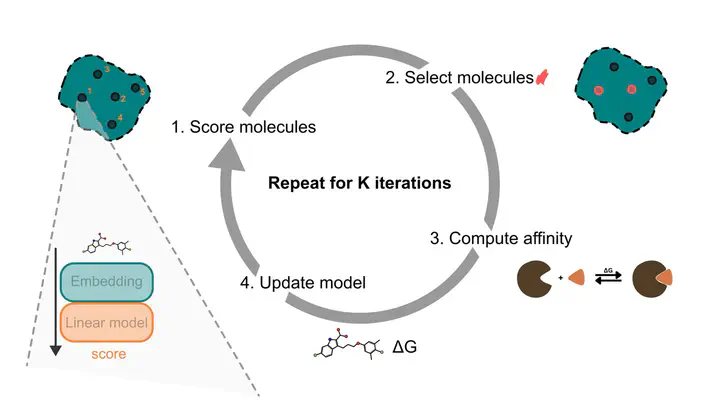
Predicting protein–ligand binding affinity with high accuracy is critical in structure-based drug discovery. While docking methods offer computational efficiency, they often lack the precision required for reliable affinity ranking. In contrast, molecular dynamics (MD)-based approaches such as MMGBSA provide more accurate binding free energy estimates but are computationally intensive, limiting their scalability. To address this trade-off, we introduce an active learning framework that automates molecule selection for docking and MD simulations, replacing manual expert-driven decisions with a data-efficient, model-guided strategy. Our approach integrates fixed, partly pre-trained deep learning molecular embeddings (MolFormer, ChemBERTa-2, and Morgan fingerprints) with adaptive regression models (e.g. Bayesian Ridge and Random Forest) to iteratively improve binding affinity predictions. We evaluate this approach retrospectively on a new dataset of 59 356 chemically diverse compounds from ZINC-22 targeting the MCL1 protein using both AutoDock Vina and MMGBSA binding free energy scores. Validation against a subset of experimentally measured binding affinities demonstrates that MMGBSA scores exhibit a stronger ranking correlation than the docking scores. Our results show that incorporating MMGBSA scores into the active learning loop enables highly efficient compound selection, recovering 79.9% of the top 1% MMGBSA-ranked binders while screening only a fraction of the dataset. In contrast, docking-guided selection identifies a largely distinct set of compounds, recovering only 6.7% of these top MMGBSA-ranked binders, underscoring the critical impact of scoring function choice. Furthermore, we demonstrate that a one-at-a-time acquisition active learning strategy consistently outperforms traditional batched acquisition, with the latter achieving just 78.4% recovery with MolFormer and Bayesian Ridge. These findings underscore the potential of integrating deep learning-based molecular representations with MD-level accuracy in an active learning framework, offering a scalable and efficient path to accelerate virtual screening and improve hit identification in drug discovery.