Computational Biophysics Group
Welcome to the Computational Biophysics Group at Saarland University.
We develop methods related to molecular dynamics simulations, with the aim to understand the relationship between structure, dynamics, and function of biological macromolecules.
Temporary contact: Aouregan Lemée

We have several interesting Bachelor and Master projects available. Find out more.
We do not currently have any open positions. However, we are always interested in hearing from ambitious PhD candidates or postdocs who are willing to apply for their own funding. Find out more.
Research Topics
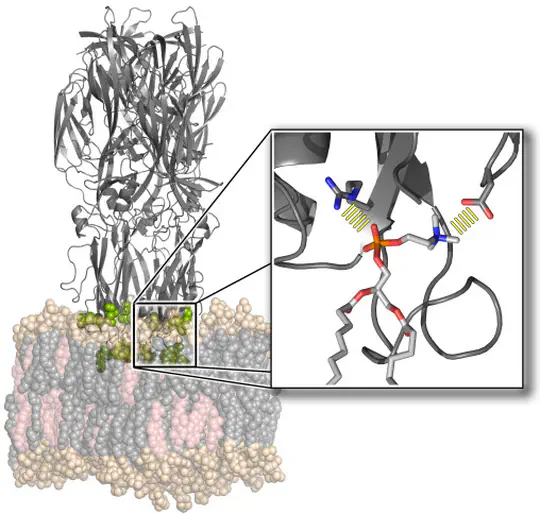
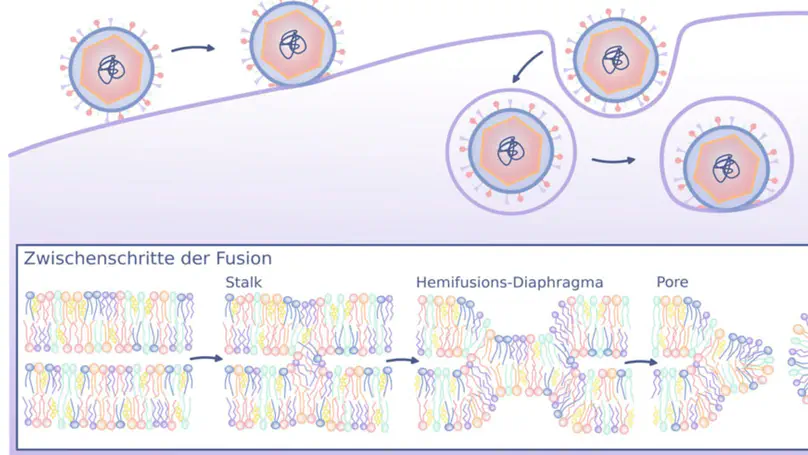
The function of biological membranes goes far beyond the formation of a mere barrier. Membranes are subject to ongoing structural remodeling, which is controlled by interactions with proteins and by the lipid composition. We develop free energy calculation techniques to understand how membrane composition and interactions with proteins (such as viral fusion proteins) enable functionally important events at membranes including membrane fusion, pore formation, or drug permeation.
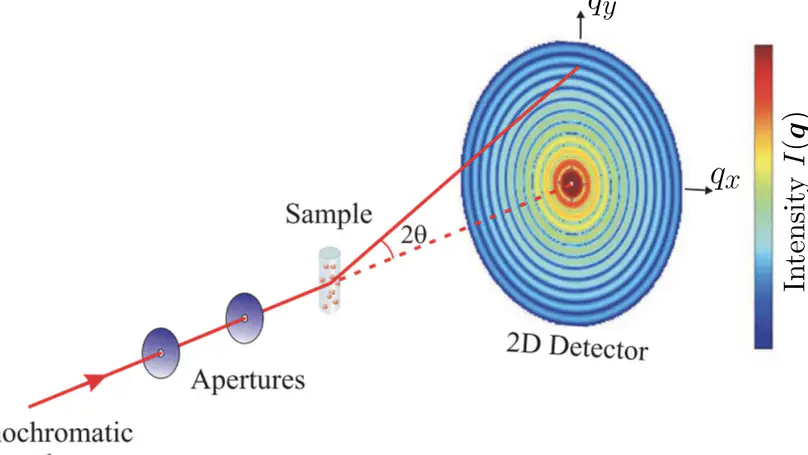
Collecting experimental data is often difficult – but the interpretation of the data may be even more challenging, for instance because the information content of the experimental signals is low. We develop methods for combining MD simulations with experimental data to get the best of two worlds, with some focus on small-angle X-ray and neutron scattering data (SAXS/SANS). Our developments involve accurate SAXS/SANS predictions, protein structure and ensemble refinement, studies on the protein hydration shell, and modeling of experiments at X-ray free electron lasers. We share our methods via the web server WAXSiS and GROMACS-SWAXS.
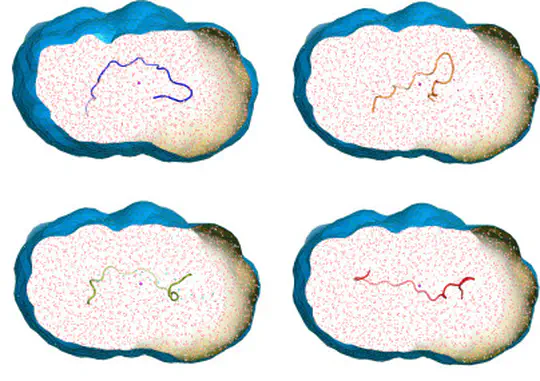
Proteins are not static building blocks but instead carry out their function –and malfunction– by structural transitions (Structure-function-dynamics relationship). We combine MD simulations with experiential data and enhanced-sampling techniques, to observe proteins while they function in atomic detail. Our portfolio comprises studies of molecular motors, protein-RNA/DNA complexes, membrane channels, and enzymes related to cancer progression.

Latest News



Latest Publications
Meet the Team
Noora Aho
Postdoc
Leonie Chatzimagas
PhD student
Joel Chavarria Rivera
Master student
Noah Garber
PhD student
Jules Grollier
PhD student
Jochen Hub
Professor of Computational Biophysics
Bettina Lau
Secretary (on leave)
Aouregan Lemée
Secretary
Kristian Lytje
Postdoc
Alejandro Martínez León
PhD student
Katharina Scherer
PhD student
Funding
Present and former