Computational Biophysics Group
Welcome to the Computational Biophysics Group at Saarland University.
We develop methods related to molecular dynamics simulations, with the aim to understand the relationship between structure, dynamics, and function of biological macromolecules.

We are hiring: a PhD position is available in computational membrane biophysics. Please apply until Feb. 9, 2026. Find out more.
We have several interesting Bachelor and Master projects available. Find out more.
Research Topics
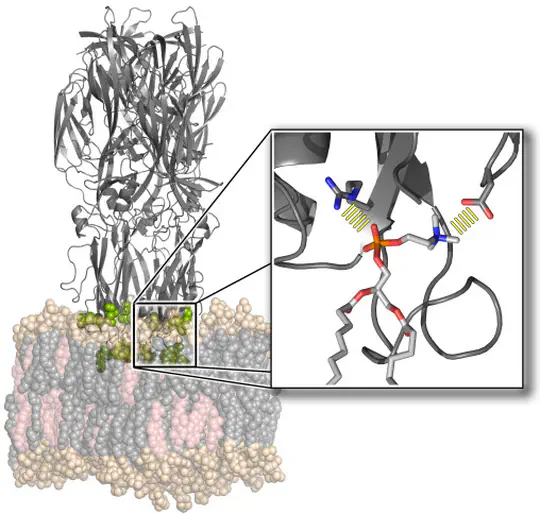
The function of biological membranes goes far beyond the formation of a mere barrier. Membranes are subject to ongoing structural remodeling, which is controlled by interactions with proteins and by the lipid composition. We develop free energy calculation techniques to understand how membrane composition and interactions with proteins (such as viral fusion proteins) enable functionally important events at membranes including membrane fusion, pore formation, or drug permeation.
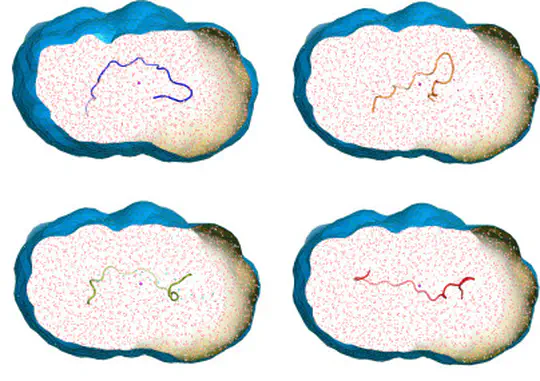
Collecting experimental data is often difficult – but the interpretation of the data may be even more challenging, for instance because the information content of the experimental signals is low. We develop methods for combining MD simulations with experimental data to get the best of two worlds, with some focus on small-angle X-ray and neutron scattering data (SAXS/SANS). Our developments involve accurate SAXS/SANS predictions, protein structure and ensemble refinement, studies on the protein hydration shell, and modeling of experiments at X-ray free electron lasers. We share our methods via the web server WAXSiS and GROMACS-SWAXS.
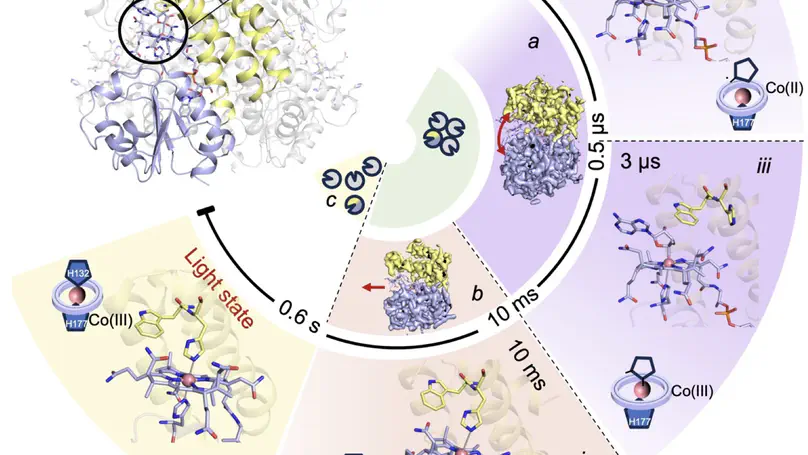
Proteins are not static building blocks but instead carry out their function –and malfunction– by structural transitions (Structure-function-dynamics relationship). We combine MD simulations with experiential data and enhanced-sampling techniques, to observe proteins while they function in atomic detail. Our portfolio comprises studies of molecular motors, protein-RNA/DNA complexes, membrane channels, and enzymes related to cancer progression.

Latest News



Latest Publications

We present BindFlow, a Python-based software for automated absolute binding free energy (ABFE) calculations at the free energy perturbation (FEP) or at the molecular mechanics Poisson–Boltzmann/generalized Born surface area [MM(PB/GB)SA] level of theory. BindFlow is free, open-source, user-friendly, and easily customizable, runs on workstations or distributed computing platforms, and provides extensive documentation and tutorials. BindFlow uses GROMACS as a molecular dynamics engine and provides built-in support for the small-molecule force fields GAFF, OpenFF, and Espaloma, as well as support for user-provided custom force fields. We test BindFlow by computing affinities for 139 receptor–ligand pairs, involving eight different targets, including six soluble proteins, one membrane protein, and one nonprotein host–guest system. We find that the agreement of BindFlow predictions with experiments is overall similar to gold standards in the field. Interestingly, we find that MM(PB/GB)SA achieves correlations that, for some systems and force fields, approach those obtained with FEP while requiring only a fraction of the computational cost. This study establishes BindFlow as a validated and accessible tool for ABFE calculations.
Meet the Team
Noora Aho
Postdoc
Leonie Chatzimagas
PhD student
Joel Chavarria Rivera
Master student
Noah Garber
PhD student
Jochen Hub
Professor of Computational Biophysics
Bettina Lau
Secretary
Johanna Linse
PhD student
Kristian Lytje
Postdoc
Alejandro Martínez León
PhD student
Katharina Scherer
PhD student
Funding
Present and former